

Генетический дефект липопротеиновой липазы (ГДЛЛ) является одной из самых распространенных наследственных дислипидемий, которая приводит к нарушению метаболизма липидов в организме. ГДЛЛ относится к группе наследственных расстройств обмена липидов и проявляется низким уровнем липазы и повышением общего количества триглицеридов и холестерола в крови.
При ГДЛЛ нарушается диагностических ферментной системы, что приводит к формированию выраженного lipidol. Одной из самых распространенными проявлений ГДЛЛ является хронический панкреатит, который часто проявляется в детском возрасте, а иногда и взрослыми после 40 лет.
Одну из основных причин развития ГДЛЛ можно назвать генетическое наследование, при котором нарушается вольмана лет в печеночно-клеточной оболочке, что ведет к формированию склероза и фиброза. Этот генотип характерен для гомозиготной формы ГДЛЛ, при которой дети наследуют болезнь от обоих родителей.
Симптомы ГДЛЛ довольно характерны и похожи на проявления других наследственных болезней обмена липидов. Они могут включать в себя увеличение уровня липидов в крови, различные кровотечения, атеросклероз, склерозирование артерий и другие органы-мишени в организме. Кроме того, пациенты с ГДЛЛ часто имеют измененные функции печени и других внутренних органов.
Причины генетического дефекта липопротеиновой липазы
Причина генетического дефекта ЛПП связана с мутациями в генах, ответственных за синтез и образование функциональных оболочек ЛПП. Эти мутации могут быть унаследованы от родителей или возникать новыми мутациями в генах.
Повышение частоты генетического дефекта ЛПП наблюдается в семьях, где ранее были случаи гиперхолестеринемии или других нарушений обмена липидов. Согласно исследованиям, причиной генетического дефекта может быть наличие нескольких мутаций в генах, отвечающих за синтез ЛПП.
Диагностика генетического дефекта ЛПП проводится с помощью различных исследований, таких как липидограммы, тесты с определением активности ЛПП и проверка наличия мутаций в генах, связанных с ЛПП. Рекомендуется проводить обследование всех пациентов с гиперхолестеринемией или подозрением на нарушение обмена липидов.
Клиническая картина и симптомы
У пациентов с генетическим дефектом ЛПП наблюдаются следующие симптомы:
- Повышенная чувствительность к холоду и боли в животе;
- Развитие фиброза и образование жировых клеток (аденомы) на языке и других органах;
- Повышенная частота развития гиперхолестеринемии и атеросклеротических изменений сосудов;
- Может развиться синдром Шардона, который характеризуется повышением уровня ЛПНП в плазме крови и наличием гетерофибратомикронов среднего типа в липоидном ядре клеток;
- Другими последствиями могут быть нарушения обмена жирных кислот, повышение уровня глюкозы в крови и т. д.
Методы лечения и терапия
Лечение генетического дефекта ЛПП направлено на снижение уровня ХС-ЛПНП и ХС-ЛПОНП в крови. Основным препаратом, используемым в терапии, является рекомбинантная ЛПП (ркнПК), который компенсирует недостаточную активность энзима ЛПП.
Также рекомендуется проведение диетотерапии с ограничением потребления насыщенных жиров и углеводов, которые могут усиливать нарушение обмена липидов. При высокой эффективности терапии возможно снижение уровня ХС-ЛПНП и ХС-ЛПОНП, а также нормализация липидной гомеостаза.
Всех пациентов с генетическим дефектом ЛПП рекомендуется периодически проходить обследования и следить за уровнем липопротеинов и жиров в крови. Также важным аспектом является обучение пациентов о возможных осложнениях и способах предотвращения их развития.
Симптомы генетического дефекта липопротеиновой липазы
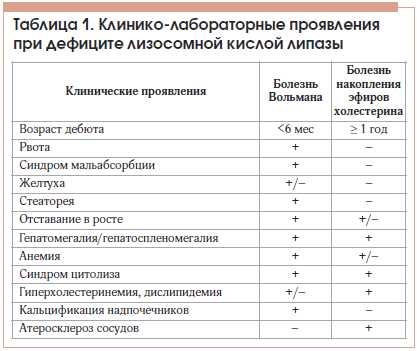
Симптомы генетического дефекта липопротеиновой липазы могут проявиться в раннем возрасте, у детей или взрослых, и могут варьироваться в зависимости от степени дефекта. Они могут включать:
- Снижение жиров в крови;
- Недостаточность усвоения пищи, содержащей жиры;
- Повышенная чувствительность к жирной пище;
- Раннее развитие атеросклероза;
- Повышенная потребность в физической активности;
- Образование жировых пробков в межтканевом пространстве;
- Набор лишнего веса, особенно в области живота и поясницы;
- Повышенное содержание триглицеридов в крови;
- Аномалии в работе поджелудочной железы и надпочечниковой железы;
- Диабет;
- Сердечно-сосудистые заболевания.
Для диагностики генетического дефекта липопротеиновой липазы необходимо проведение генетических тестов, включая анализ мутаций в генах, отвечающих за синтез липазы. Кроме того, лечение этого генетического дефекта может включать правильный подход к питанию, физическую активность и медикаментозную терапию.
Взаимодействие с врачом и соблюдение рекомендаций важны для снижения риска осложнений и улучшения качества жизни пациентов. Регулярные тесты на уровень липидов в крови и обследования, такие как томография и другие методы изображения, также могут быть важной частью диагноза и лечебного процесса.
Методы диагностики генетического дефекта липопротеиновой липазы
Молекулярная генетика:
Для постановки диагноза генетического дефекта липопротеиновой липазы проводят молекулярно-генетическое исследование. При этом ищут специфические мутации гена, ответственного за этот дефект. Мутации в гене LPL могут быть гетерозиготными или гомозиготными.
Клиническое обследование:
При клиническом обследовании пациента врач может выявить следующие симптомы генетического дефекта липопротеиновой липазы: нарушение функции поджелудочной железы, нарушение обмена жирных кислот, повышение содержания жирных кислот в крови, а также наличие других осложнений.
Лабораторные методы:
Для подтверждения диагноза генетического дефекта липопротеиновой липазы используются следующие лабораторные методы:
- Липидограммы — анализ, позволяющий оценить количество липопротеинов и их состав в крови.
- Аполипопротеинограммы — анализ, который позволяет определить количество и состав аполипопротеинов в крови.
- Холестерин-ЛПВП — анализ, который позволяет выявить количество холестерина в липопротеинах низкой плотности (ЛПНП).
- Эластографии — метод, использующийся для оценки эластичности и функции разных органов-мишеней при генетическом дефекте липопротеиновой липазы.
Для диагностики генетического дефекта липопротеиновой липазы также рекомендуется проведение доказательной терапии и детского обследования, чтобы выявить возможные осложнения и следить за функцией разных органов-мишеней. Вопросы генетики и молекулярной генетики являются ключевыми в этом.
Методы лечения генетического дефекта липопротеиновой липазы
Лечение генетического дефекта липопротеиновой липазы основано на комплексном подходе и включает использование лекарственных препаратов, диеты и других методов.
Лекарственные препараты
Один из основных методов лечения генетического дефекта липопротеиновой липазы — применение лекарственных препаратов для снижения уровня жирных кислот и триглицеридов в крови. Рекомендуется проконсультироваться с врачом, чтобы составить правильный план терапии с учетом индивидуальных особенностей пациента и степени проявления дефекта.
Одним из препаратов, которые могут быть приведены в научной и клинической практике, являются статины, которые способствуют снижению уровня холестерина в крови. Однако выбор лекарственного препарата и его дозировка должны быть определены врачом.
Диета и изменение образа жизни
Специалисты рекомендуют включить в комплекс лечения генетического дефекта липопротеиновой липазы правильное питание и регулярные физические нагрузки. Важно следовать рекомендациям специалиста по составлению диеты, которая включает ограничение потребления жиров, особенно насыщенных, и привлечение продуктов, богатых полиненасыщенными жирными кислотами. Это может включать рыбу, орехи, семена и другие источники полезных жиров.
Также рекомендуется вести активный образ жизни и включать в режим физическую активность, по возможности каждые 30 минут в день.
Дополнительные методы лечения
В некоторых случаях может потребоваться применение дополнительных методов лечения. Например, для контроля уровня жиров в крови может быть назначено приложение медицинского прибора. Также могут быть использованы другие методы, такие как аланинаминотрансферазы и аполипопротеинограммы, для более точного анализа состояния пациента.
В случае тяжелого генетического дефекта липопротеиновой липазы, когда нарушения обмена жирных кислот размером с мозга проявляются уже у детей и сопровождаются гиперхолестеринемии, дислипидемия, стеатореей, фиброзом печени или даже нимана-пика можем быть привлечены редкие методы лечения, проведенные экспертами в области генетики и педиатрии.
В целом, вопросы лечения генетического дефекта липопротеиновой липазы требуют дальнейших научных исследований, и мнение специалистов может меняться по мере расширения знаний об этой патологии.
Прогноз и осложнения при генетическом дефекте липопротеиновой липазы
Эпидемиологические исследования показывают, что частота ГДЛЛ в России составляет примерно 1 на 1 миллион живорожденных. В семье пациентов с ГДЛЛ риск появления этого заболевания у других членов семьи составляет 25%. Несмотря на свою редкость, ГДЛЛ отличается высокими клиническими проявлениями и тяжестью течения. Симптомы ГДЛЛ могут быть похожи на другие заболевания с нарушением усвоения жиров и могут включать стеаторею, гиперчувствительность к холестерину, недостаточность активности аланинаминотрансферазы.
Дефицит липопротеиновой липазы приводит к нарушению расщепления триглицеридов и усвоению кислоты. Это может вызывать накопление жиров в тканях и органах, что в свою очередь приводит к образованию ксантом и нимана-пика, а также повышению уровня холестерина и триглицеридов в крови. Поэтому пациенты с ГДЛЛ испытывают ряд осложнений, включая сердечно-сосудистые заболевания, панкреатит, гепатоспленомегалию и диабет.
Прогноз при ГДЛЛ зависит от своевременного выявления и правильного лечения. Современные методы лечения включают заместительную терапию липопротеиновой липазы, контроль пищевого рациона и физическую активность. Специалисты в области медицины активно занимаются исследованиями в этой области и постоянно разрабатывают новые методики лечения и улучшение прогноза для пациентов с ГДЛЛ.
Таким образом, генетический дефект липопротеиновой липазы является очень редким и тяжелым заболеванием, которое может вызывать серьезные осложнения в разных органах и системах организма пациента. Подходящее лечение и контроль над питанием и физической активностью могут помочь в управлении этим заболеванием и улучшении прогноза для пациентов.
Классификация генетического дефекта липопротеиновой липазы
Вопросы классификации генетического дефекта липопротеиновой липазы включают в себя эпидемиологические, генетические, физиологические и биохимические аспекты данного расстройства обмена веществ.
Основная классификация ГДЛЛ:
- ГДЛЛ первого типа (ГДЛЛ1): Генетический дефект липопротеиновой липазы;
- ГДЛЛ второго типа (ГДЛЛ2): Дефект аполипопротеина CII или аполипопротеина CIII;
- ГДЛЛ третьего типа (ГДЛЛ3): Дефект аполипопротеина E;
- ГДЛЛ четвертого типа (ГДЛЛ4): Дефект ангиопоэтина-липопротеина;
- ГДЛЛ пятого типа (ГДЛЛ5): Дефект липопротеинлипазного механизма.
Этническая переменчивость и генетические особенности также играют роль в классификации ГДЛЛ. Например, ГДЛЛ1 включает форму, характеризующуюся наличием эндогенного ингибитора липопротеиновой липазы ткань-специфического ингибитора (ТСИ).
Другие классификационные критерии:

- Клинического подхода: классификация основывается на клинической презентации и особенностях пациентов с ГДЛЛ;
- Классификация по преобладающим симптомам и осложнениям: одним из подходов может быть классификация по наличию стеатореи (жирная диарея) и других осложнений;
- Классификация по биомаркерам: основывается на наличии и количестве биомаркеров, таких как холестерол, триглицериды и другие;
- Классификация научных исследований: отражает последние достижения в области исследования ГДЛЛ и ее подтипов;
- Классификация по наличию в клинических рекомендациях: основывается на наличии рекомендаций по диагностике и лечению ГДЛЛ в различных специальностях, таких как гастроэнтерология, общая медицина и др.
Изучение генетического дефекта липопротеиновой липазы и его классификация играют важную роль в определении причин, симптомов и методов лечения этого нарушения обмена веществ.
0 Комментариев